
Large ED-based trial finds no benefit for azithromycin in preschoolers, even when pathogenic bacteria are present
The issue: Up to one-quarter of preschool wheezing episodes in the U.S. receive antibiotic therapy, often azithromycin, despite wheezing being predominantly viral in origin.
Why this study matters: The AZ-SWED trial randomized 840 preschoolers presenting to the ED with moderate-to-severe wheezing to azithromycin or placebo and found no difference in symptom severity, even in children colonized with pathogenic bacteria.
Primary care relevance: Reinforces that azithromycin should not be used routinely for wheezing in preschoolers presenting to the ED.
SURMOUNT-MAINTAIN evaluated continuing, reducing, or stopping Zepbound after initial weight loss
The issue: Patients who reach their weight-loss goal on Zepbound often ask what maintenance therapy should look like.
Why this study matters: SURMOUNT-MAINTAIN randomized adults after 60 weeks of tirzepatide to continue maximum dose, reduce to 5 mg, or switch to placebo for 52 weeks.
Primary care relevance: Provides data to counsel patients on the maintenance phase of weight loss with tirzepatide.

ATTAIN-MAINTAIN trial supports switching to Foundayo after Wegovy or Zepbound
The issue: Patients who reach their weight-loss goal on injectable GLP therapies often ask what their maintenance therapy options are.
Why this study matters: It evaluated the effects of switching to Foundayo after injectable Wegovy or Zepbound.
Primary care relevance: Provides data on counseling patients who want to switch to oral GLP therapy after injections.

A small, placebo-controlled trial showed that semaglutide reduced heavy drinking days in obese patients
The issue: Alcohol use disorder (AUD) is common but lacks a highly effective treatment.
Why this study matters: It evaluated the effects of semaglutide on drinking habits in adults with AUD and obesity.
Primary care relevance: This study suggests semaglutide may help curb drinking in obese patients with AUD.

Studies provide insight into exercise-induced hypoglycemia
The issue: Fear of exercise-induced hypoglycemia remains a common barrier to physical activity in type 1 diabetes, and clinicians need practical data to counsel patients.
Why these studies matter: They report real-world glucose changes across hundreds of thousands of exercise sessions and introduce a three-variable heatmap (GlucoseGo) to estimate hypoglycemia risk before exercise.
Primary care relevance: Provides guidance on counseling type 1 diabetics about exercise
The PANORAMIC trial did not find Paxlovid to be beneficial in a vaccinated population
The issue: The effects of Paxlovid on significant clinical outcomes in vaccinated adults have not been evaluated in a large trial
Why this study matters: It evaluated the effects of Paxlovid on hospitalizations and death in a mostly vaccinated (98%) population
Primary care relevance: Providers can reassure most vaccinated patients that Paxlovid is unnecessary
Trial found modest symptom improvement but increased bleeding risk with venous stenting
The issue: Post-thrombotic syndrome (PTS) can cause chronic limb symptoms after DVT. Effective PTS therapies are lacking.
Why this study matters: Evaluated the effects of iliac vein stent placement for PTS
Primary care relevance: Offers guidance on counseling patients with PTS who are considering endovascular therapy

Randomized trial reveals common conservative treatment for CTS may not provide significant clinical benefit over placebo
The issue: Wrist splinting is a common first-line treatment for carpal tunnel syndrome, but evidence supporting its efficacy is limited.
Why this study matters: In a randomized trial (N=142), symptom improvement at 12 weeks and surgery rates at 1 year were similar for rigid wrist splinting and a placebo soft bandage.
Primary care relevance: Helps set expectations for conservative care and may support earlier discussion of alternative treatments in patients with persistent symptoms.
One-quarter of adults aged 60 and older successfully discontinued thyroid replacement therapy in a prospective cohort study
The issue: Levothyroxine is often continued for life, but many older adults may be taking it without a permanent indication.
Why this matters: In a prospective study of 370 adults aged 60 and older, 25.7% discontinued levothyroxine while maintaining acceptable thyroid function at 1 year.
Primary care relevance: Supports supervised dose reduction in selected patients, particularly those on lower baseline doses.
Once-weekly Awiqli (insulin icodec-abae) is expected in pharmacies in the second half of 2026
The issue: The FDA recently approved the first once-weekly insulin for type 2 diabetes
Why this matters: Once-weekly insulin may appeal to patients who do not like daily injections
Primary care relevance: Providers need to be aware of Awiqli's effects and dosing
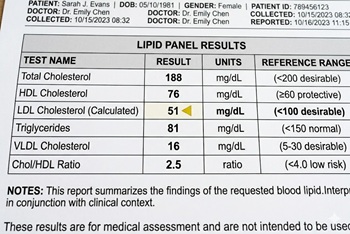
The Ez-PAVE trial randomized patients with cardiovascular disease (CVD) to an LDL target of less than 55 mg/dL versus less than 70 mg/dL
The issue: Recent American Heart Association (AHA) cholesterol treatment guidelines include an optional, stricter LDL target of less than 55 mg/dl for patients at very high risk for recurrent cardiovascular events. Studies evaluating target levels this low are lacking.
Why this study matters: The Ez-PAVE trial evaluated the effects of an LDL target below 55 mg/dL versus below 70 mg/dL in patients with CVD
Primary care relevance: The trial provides clinical evidence on the effects of targeting very low LDL cholesterol levels
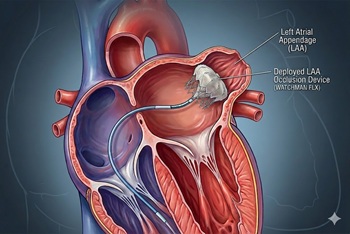
Unlike previous trials, LAAC did not perform as well in the CLOSURE-AF trial, which enrolled high-risk patients
The issue: Left atrial appendage closure (LAAC) has been viewed as an alternative to long-term anticoagulation in atrial fibrillation, particularly when bleeding risk is high.
Why this study matters: The CLOSURE-AF trial randomized patients at high risk for stroke and bleeding to LAAC or physician-directed therapy (mostly DOACs); LAAC was not noninferior for the primary composite endpoint over a median of 3 years.
Primary care relevance: Reinforces that LAAC is not a one-size-fits-all substitute for anticoagulation in the highest-risk multimorbid patients; perioperative antithrombotic needs and complications matter in shared decision-making.
Results were likely biased by the Hawthorne effect
The issue: Increased fluid intake is universally recommended to help prevent recurrent kidney stones, but adherence is difficult.
Why this study matters: In the PUSH trial, an intensive adherence intervention increased urine volume only modestly and did not reduce symptomatic stone recurrence over two years.
Primary care relevance: Reinforcing hydration remains reasonable, but clinicians should recognize that achieving >2.5 L/day urine output may be unrealistic for many patients.
Foundayo is the second oral GLP-1 drug approved for weight management
The issue: GLP-1 weight-loss therapies are the most talked-about drug class ever
Why this matters: In January 2026, Wegovy tablets became the first oral GLP-1 drug approved for weight loss. Now, a second option, Foundayo, is available.
Primary care relevance: Providers should be aware of the differences in efficacy, cost, and convenience of available therapies
Randomized trial finds holding one preprocedure dose of a GLP-1 analog reduces residual gastric volume
The issue: GLP-1 analogs are being increasingly prescribed for weight loss and type 2 diabetes, but their gastric-slowing effects have raised concerns about residual gastric volume (RGV) and potential pulmonary aspiration during procedures requiring sedation.
Why this study matters: It compared residual gastric volumes (RGV) during endoscopy in patients who held one preprocedure GLP-1 analog dose to those who did not.
Primary care relevance: Primary care providers are often asked about holding medications before procedures. This study provides some objective evidence that holding GLP-1 analogs for a single dose before procedures reduces RGV.
COBBRA trial shows lower bleeding risk with apixaban over 3 months without increased recurrent VTE
The issue: Apixaban and rivaroxaban are both widely used for acute VTE, but large randomized trials comparing their effects are lacking
Why this study matters: The COBBRA trial compared apixaban to rivaroxaban for the treatment of acute symptomatic venous thromboembolism
Primary care relevance: Study provides comparative evidence on apixaban and rivaroxaban for acute VTE
The 2026 update is the first significant update in eight years, replacing the 2018 guidelines
The issue: Lipid therapies have evolved significantly since 2018, and large studies evaluating their effects have been published
Why this matters: The 2026 guidelines endorse more aggressive screening and treatment goals
Primary care relevance: Treating high cholesterol is a key component of primary care, and clinicians should be aware of the new guidelines and how to implement them
Exact Sciences expands into blood-based multi-cancer early detection with Cancerguard
The issue: Early detection for many cancers remains limited because no routine screening tests exist for most malignancies.
Why this matters: Cancerguard is a blood-based multi-cancer early detection test that claims to detect more than 50 cancer types and subtypes.
Primary care relevance: Exact Sciences is advertising Cancerguard, and patients may have questions about it. Providers should be aware of the test and its limitations.
Intranasal etripamil (Cardamyst) offers a new self-administered option for rapid PSVT conversion in select patients
The issue: Vagal maneuvers are first-line for stable acute SVT, but real-world conversion rates range from 5 to 20%, and many patients still seek emergency care.
Why this matters: In its pivotal trial, Cardamyst (etripamil) converted AV-nodal-dependent PSVT within 30 minutes in 64% of patients vs 31% with placebo
Primary care relevance: Cardamyst provides an at-home, symptom-prompted option for selected adults who want episodic control of SVT, possibly decreasing emergency department visits and deferring catheter ablation.
The new 7.2 mg dose is triple the previous highest dose of 2.4 mg
The issue: Average weight loss with the highest dose of Wegovy is 13% to 16%, while its main competitor, Zepbound, has a mean weight loss of 20% to 22% at its highest dose
Why this matters: A study comparing Wegovy doses of 2.4 mg and 7.2 mg found that the higher dose caused more weight loss, but also more gastrointestinal side effects and dysaesthesia.
Primary care relevance: Providers will soon be able to prescribe the higher dose, but should counsel patients about the potential for more gastrointestinal side effects and dysaesthesia.
Trial finds 5% permethrin cream superior to oral ivermectin for scabies in children and adults
The issue: Scabies is a common skin infection caused by a parasitic mite. It is typically treated with topical medications, but the best treatment is uncertain.
Why this study matters: It compared topical permethrin to oral ivermectin for scabies in children and adults.
Primary care relevance: Supports permethrin as first-line treatment for scabies. Provides cure rates for permethrin and ivermectin.
STABLED trial found no benefit of adding catheter ablation to edoxaban in patients with AF and recent stroke
The issue: Catheter ablation is widely used for atrial fibrillation; whether adding it to anticoagulation improves hard outcomes in high-risk patients, such as those with a recent stroke, is uncertain.
Why this study matters: It evaluated the safety and efficacy of adding catheter ablation to anticoagulation in patients with AF and recent stroke.
Primary care relevance: Provides information for counseling patients about the benefits and risks of catheter ablation.
Population-based MRI study finds rotator cuff abnormalities in nearly all adults over 40, with poor correlation to symptoms
The issue: Shoulder pain is a common complaint, and imaging is frequently ordered
Why this study matters: It evaluated the prevalence of rotator cuff abnormalities on MRI in the general population and found that they are common in asymptomatic people.
Primary care relevance: Highlights the hazard of over-imaging: incidental findings are common in asymptomatic people and may lead to unnecessary treatment or surgery; routine MRI for atraumatic shoulder pain has limited value.

Individual-patient-data meta-analysis finds no benefit of beta-blockers after MI in patients with preserved ejection fraction
The issue: Four large trials evaluating the benefits of beta-blockers in post-MI patients with preserved LVEF came to different conclusions
Why this study matters: It combines data from the trials and offers an estimate of the overall effect of beta-blockers in this patient population.
Primary care relevance: Supports reassessing the need for long-term beta-blockers in post-MI patients with preserved EF.

Tradipitant (Nereus®), an NK-1 receptor antagonist, is the first drug in its class approved for the prevention of motion sickness
The issue: Motion sickness is a common problem that affects many people, especially travelers.
Why this matters: Tradipitant (Nereus®) is a new drug for motion sickness prevention with a novel mechanism of action.
Primary care relevance: Offers a new prescription option for motion sickness prevention with a different mechanism than scopolamine or antihistamines.

Randomized trial finds 15% dose increase when taking levothyroxine with breakfast maintains TSH stability
The issue: Food reduces the absorption of levothyroxine (LT4); therefore, dosing guidelines recommend taking it on an empty stomach, typically 30 to 60 minutes before breakfast. Many patients find this inconvenient.
Why this study matters: It randomized 88 adults with hypothyroidism to fasting LT4 intake or LT4 intake with breakfast with a 15% dose increase. The primary outcome, TSH stability, did not differ significantly between groups.
Primary care relevance: For patients who prefer taking LT4 with food, this study provides guidance on dose adjustment.
The vaccine had a significant effect but no meaningful overall benefit
The issue: Several RSV vaccines for adults have been developed in recent years and are heavily marketed to seniors, but data on their real-world effectiveness against hospitalization are limited.
Why this study matters: The DAN-RSV open-label trial randomized 131,276 adults 60 years and older to RSVpreF vaccine (Abrysvo®) or no vaccine; hospitalization for RSV-related respiratory tract disease occurred in 3 vs. 18 participants (vaccine effectiveness, 83.3%; P=0.007).
Primary care relevance: The study primarily showed that RSV does not appear to be a significant cause of respiratory illness in older adults; the NNT to prevent one RSV hospitalization was approximately 4400, indicating minimal individual benefit.
PISCES trial finds fish oil reduces cardiovascular events in hemodialysis patients
The issue: Large trials of fish oil for CVD prevention have yielded conflicting results; REDUCE-IT showed benefit while STRENGTH did not.
Why this study matters: The PISCES trial randomized 1,228 hemodialysis patients to fish oil 4 g daily or corn-oil placebo; fish oil reduced serious cardiovascular events by 43% (HR 0.57; 95% CI, 0.47 to 0.70; P<0.001) over 3.5 years.
Primary care relevance: PISCES aligns with REDUCE-IT but contradicts STRENGTH; the overall effects of fish oil on CVD remain inconclusive.
ESCUDDO trial finds single-dose HPV vaccination noninferior to two doses
The issue: Current guidelines recommend two doses of Gardasil 9 for adolescents; whether one dose provides comparable protection has been uncertain.
Why this study matters: The ESCUDDO trial randomized girls 12 to 16 years to one or two doses of bivalent (Cervarix) or nonavalent (Gardasil 9) HPV vaccine; one dose was noninferior to two doses for preventing HPV16 or HPV18 infection.
Primary care relevance: Supports WHO single-dose recommendation; study has limitations including 5-year follow-up and nonrandomized control group requiring propensity matching.
Recent studies show benefit in severe disease, but routine use remains unproven
The issue: Studies evaluating corticosteroids in CAP have produced mixed results; their role in routine practice is uncertain.
Why these studies matter: The SONIA trial showed reduced mortality with glucocorticoids in low-resource settings; the CAPE COD trial showed benefit in ICU patients.
Primary care relevance: The IDSA recommends against routine steroids in CAP; evidence supports use only in severely ill patients requiring ICU care.

Self-collection is convenient and time-saving for many women, but only one option exists for home collection
The issue: Cervical cancer screening has traditionally required an in-person visit and pelvic exam; that barrier contributes to underscreening, especially among people who lack access to gynecologic care or have difficulty scheduling visits.
Primary care relevance: Provides guidance on self-collected swabs for cervical cancer screening.
Retreatment reduces viral loads and symptoms by one day but shows no meaningful benefit on hospitalizations or deaths
The issue: Some patients experience COVID symptom rebound after completing Paxlovid; the safety and benefit of retreatment have not been studied.
Why this study matters: This study evaluated the safety and benefits of a second course of Paxlovid in patients with rebound COVID symptoms.
Primary care relevance: Provides guidance on whether to retreat COVID-19 patients experiencing rebound symptoms.

Zoliflodacin and gepotidacin offer oral alternatives to injectable ceftriaxone
The issue: Gonorrhea resistance to current therapies is increasing.
Primary care relevance: Providers now have two new oral antibiotics to treat gonorrhea, potentially improving treatment access and patient convenience.
Two randomized trials examine whether anticoagulation can be safely discontinued after successful catheter ablation
The issue: One-year success rates of catheter ablation for atrial fibrillation range from 50 - 85% and decline over time. Many patients hope to stop anticoagulation after ablation, but the risks and benefits of this decision have not been clearly defined.
Why these studies matter: The ALONE-AF and OCEAN studies were designed to evaluate the risks and benefits of stopping anticoagulation after successful catheter ablation. They also provided guidance on what defines successful ablation.
Primary care relevance: The studies provide data for making an informed decision on stopping anticoagulation and determining which patients are candidates.
New randomized trial compares carotid stenting and endarterectomy to intensive medical management
The issue: In the U.S., most asymptomatic high-grade (≥70%) carotid stenosis is treated with revascularization (stenting or endarterectomy). However, the risks and benefits of this approach, and how it compares with contemporary medical therapy, have never been evaluated in a clinical trial.
Why this study matters: The CREST-2 study provides the first modern evidence comparing carotid stenting and endarterectomy to intensive medical management for asymptomatic carotid stenosis.
Primary care relevance: Providers now have good data to counsel patients on the risks and benefits of stenting, endarterectomy, and medical management of asymptomatic carotid stenosis.

DECAF trial challenges long-standing recommendations to avoid coffee
The issue: Patients with atrial fibrillation are routinely advised to avoid caffeinated beverages, but evidence supporting this recommendation has been inconsistent.
Why this study matters: The DECAF randomized clinical trial compared continuing caffeinated coffee consumption to abstinence in patients with atrial fibrillation who underwent successful electrical cardioversion.
Primary care relevance: Provides guidance on counseling patients with atrial fibrillation about coffee consumption.

Lynkuet joins Veozah as a nonhormonal option for vasomotor symptoms
The issue: Many women seek nonhormonal alternatives for menopausal hot flashes due to concerns about hormone replacement therapy.
Primary care relevance: Women now have two nonhormonal options for treating menopausal hot flashes, though both medications are more expensive than HRT and often require prior authorization.
Recent trials question routine long-term beta-blocker use after MI
The issue: Beta-blockers are routinely recommended for all post-MI patients, but this practice is based on trials predating modern revascularization and intensive secondary prevention.
Why this study matters: Four recent randomized trials have evaluated the long-term benefit of beta-blockers in post-MI patients with preserved LVEF.
Primary care relevance: Provides guidance on continuing beta-blockers in post-MI patients with preserved LVEF.
